
Challenges in analysing high-dimensional data

Key to a ‘systems immunology’ approach to studing inflammation and infectious disease is the capacity to analyse and integrate datasets generated by different cytometry or single-cell technologies. In particular, the challenges in analysing high-dimensional flow, spectral, or mass cytometry (CyTOF) datasets are different to those typically faced in analysing single-cell sequencing data. This is largely due to the large number of cells in cytometry datasets, consisting of tens to hundreds of millions of cells. However, fewer features can be measured on individual cells through these cytometry technologies (typically <50 proteins), when compared to single-cell multiomic approaches (such as CITE-seq: thousands of transcripts and hundreds of proteins), though fewer cells are measured.
Ideally, we could utilise a system that is versatile enough to manage data generated through different cytometry or single-cell technologies, with a built in capacity to integrate datasets across batches, experiments, or technologies.
Analysis of large cytometry and single-cell datasets with Spectre
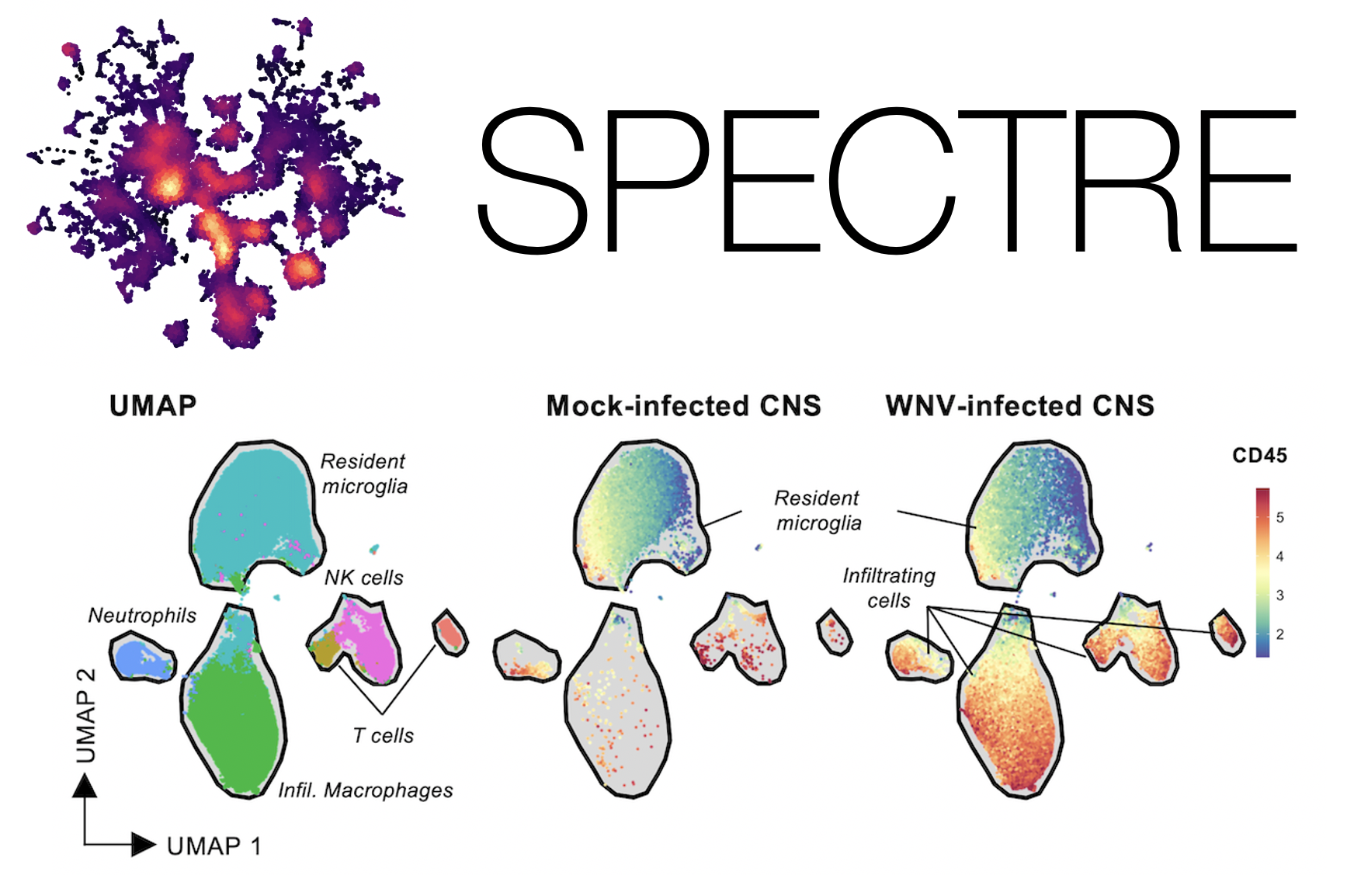
A major focus of our collaborative group is the development of computational analysis solutions for high-dimensional cytometry and single-cell data. To address the challenges listed above, we developed ‘Spectre’, a computational toolkit in R that enables comprehensive end-to-end integration, exploration, and analysis of high-dimensional cytometry data from different batches or experiments (Ashhurst et al, 2021). Spectre streamlines the analytical stages of raw data pre-processing, batch alignment, data integration, clustering, dimensionality reduction, visualisation and population labelling, as well as quantitative and statistical analysis; with a simple, clear, and modular design of analysis workflows, that can be utilised by data and laboratory scientists.
Data integration and classification
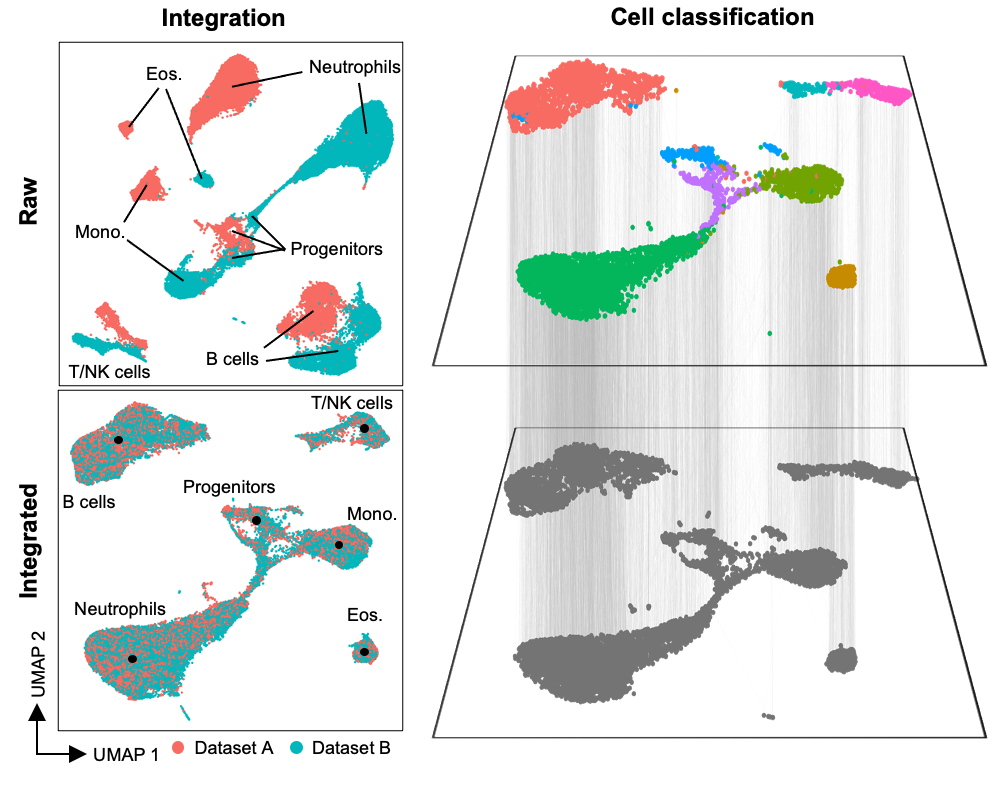
Inspired by the efforts of the Human Cell Atlas (HCA), we sought to create an interface that would allow a seamless transition across a wide variety of single-cell data types - including flow, spectral, CyTOF, and scRNAseq, including multiomic assays such as CITE-seq and Abseq. In particular, our goal was to faciliate integration and label transfer between datasets, so that reference datasets generated by the HCA and other groups could be leveraged in a wide variety of contexts, in both HD cytometry and single-cell sequencing. By creating a technology and data-agnostic platform, we can efficiently incorporate various analysis tools, and particular integration tools, developed for cytometry (e.g. FlowSOM, CytoNorm) and transcriptomics (e.g. Harmony, Liger, Seurat). By doing this, multiple approaches can be efficiently run and compared within a single environment. Critically, as many existing scRNAseq tools do not scale well to large datasets, we have focused our analysis and integration approaches around methods that can scale to large datasets consisting of millions of cells.
Strategic implementation of batch-alignment, data-integration, and cell-type classification tools allow for the integrated analysis of multiple experiments, as well as a reproducible system for rapid and repeated cell type identification in large datasets. In addition to high-dimensional cytometry datasets, we’ve also developed functions to allow for spatial analysis of high-dimensional imaging datasets, such as those generated by Imaging Mass Cytometry.